





By Molnár-Institute for…
Subscribe to Supplier
Subscribe to Supplier

QbD in pharmaceutical method development with DryLab®4
Molnar-Institute has developed its proprietary software suite, DryLab® as a revolutionary liquid chromatography method development and optimization tool that also incorporates QbD (Quality by Design) principles into the entire process.
DryLab®4 thus supports analytical scientists in UPLC, high performance and ultra-high performance liquid chromatography (HPLC/UHPLC) method development, optimization, troubleshooting, robustness testing, and ongoing quality assurance.
DryLab® thus offers considerable speed, efficiency and risk mitigation advantages to any organisation developing HPLC methods that wish to optimize complex samples.
Quality by Design (QbD) FDA guidelines
The Quality by Design (QbD) concept was first proposed by the Romanian-born engineer and management consultant Joseph M. Juran in the late 1980s. Juran originally envisaged QbD in a manufacturing context as creating features within product planning and design that create customer satisfaction and ensuring the reliability of such features. The sum of the QbD approach is thus to remove possibilities for failure.
In the pharmaceutical context, the US Food and Drug Administration (FDA) adopted Juran’s Concept and repurposed it to the manufacture of drugs and medicines, outlining FDA QbD principles in its report “Pharmaceutical Quality for the 21st Century: A Risk-Based Approach.”
Since then, the FDA has incorporated QbD principles into its pre-market processes, emphasizing quality being ‘built in’ to products with understanding of both the product and the processes by which it will be developed and manufactured along with acknowledgement of all risks involved and how best to mitigate them.
QbD ICH requirements
Working with regulators in the European Union (the European Medicines Agency) and Japan, the FDA has furthered QbD objectives through the International Conference on Harmonisation (ICH) in its Technical Requirements for Registration of Pharmaceuticals for Human Use, whose Guidelines Q8 through Q11 encapsulate unified QbD recommendations to help researchers and manufacturers implement QbD into their own processes. Guideline Q8 (revised 2008) describes QbD-based drug formulation development while Guideline Q9 describes Quality Risk Management plans, Q10 explains Pharmaceutical Quality Systems, and Q11 refers to development of active pharmacological substances including biologics.
FDA principles define QbD as “a systematic approach to development that begins with predefined objectives and emphasizes product and process understanding based on sound science and quality risk management”. To align with the FDA and ICH guidelines, method validation should follow a three-stage approach:
- Method Design: Define method requirements and conditions and identify critical controls.
- Method Qualification: Confirm that the method is capable of meeting the design intent.
- Continued Method Verification: Gain ongoing assurance to ensure that the method remains under control in routine use.
The Method Design must include creation of an Analytical Target Profile (ATP) that maps the characteristics defining suitability of the method for its intended use. These are similar to the performance characteristics described in ICH Q2 for traditional approaches, but must now be investigated in a risk assessment exercise as described in ICH Q9 combined with development studies to identify the critical method and the sources of variation.
Robustness and ruggedness experiments are then used to investigate the relationship between method input variables and the various method performance characteristics. These should be based on Design of Experiments (DoE) disciplines to ensure maximum understanding is gained while minimizing the number of experiments. Results are then compared to the desired outcome defined in the ATP. From this, it is possible to identify a set of operational method controls and define a control strategy to be built into the method to ensure consistent performance throughout its life cycle.
Using QbD in method development provides the flexibility to perform a qualification against the specific ATP defined for intended use.
DryLab®4 QbD pharma method development in UPLC/HPLC/UHPLC
The DryLab®4 software predicts chromatograms under a much wider range of experimental conditions in silico than would be possible in the laboratory.
HPLC researchers can use DryLab® to simultaneously vary multiple method parameters such as pH, temperature, buffer concentration, and ternary eluent composition (tC) to determine exactly how separations will behave.
Crucially, the DryLab® operating procedure is constructed around QbD principles, starting with definition of a chosen ATP and then relying on DryLab®4 to shape a systematic approach and initial method conditions.
The second step is to run required input experiments, uploading them as *.cdf files, the universal format for chromatographic data systems. DryLab®4 offers a number of user-friendly features to help manage input runs, streamline peak tracking, and control for errors to generate a matched peak table, ready to build the HPLC model.
Based on initial Design of Experiment (DoE), DryLab® will create multi-dimensional resolution models that depict the set of conditions to achieve baseline separation or higher. Predicted chromatograms are viewable for any point within the model, allowing selection of the working point with the highest critical resolution. From here, DryLab® can test the separation’s tolerance limits by evaluating the impact of small fluctuations on up to 10 critical separation parameters. This allows researcher to determine those parameters exerting greatest influence on the method that must therefore be strictly controlled to achieve complete success.
cGMP Documented method
Finally, the researcher can create GMP Method Documentation (a comprehensive Knowledge Management report) that pulls all method data directly from DryLab®4 to provide a platform from which to justify and comment on method criteria and choices. This report is both GMP compliant and also conforms with FDA and ICH guidelines for incorporating QbD into the UPLC/HPLC/UHPLC process.
DryLab®’s advanced functions include several QbD relevant features:
- Peak Tracking: Identification and assignment of peaks from a set of systematic experiments is an important first step in controlling the HPLC method development process. The Comparison Feature checks original experimental runs against the DryLab® model to control for errors.
- Robustness Module: This module tests the tolerance limits of selected working point by computing the number of Out-of-Specification (OoS) results that occur given small fluctuations in method parameters, evaluating variable conditions such as gradient time, and shape temperature, pH or ternary composition, flow rate and gradient start/end points.
- DryLab®4 Knowledge Management Module: DryLab®4’s documenting and archiving tool encourages a Quality by Design (QbD) approach to method development and ensures that method conforms to standard by providing a comprehensive method report, including a platform for the step-by-step justification of method choices, with automatically generated Analytical Method Summary for submissions. This assists in achieving full GxP compliance, while enabling easier and more effective collaboration between departments, supporting analytical method transfer during development and manufacturing.
Resources
Click on DryLab simulation to see video
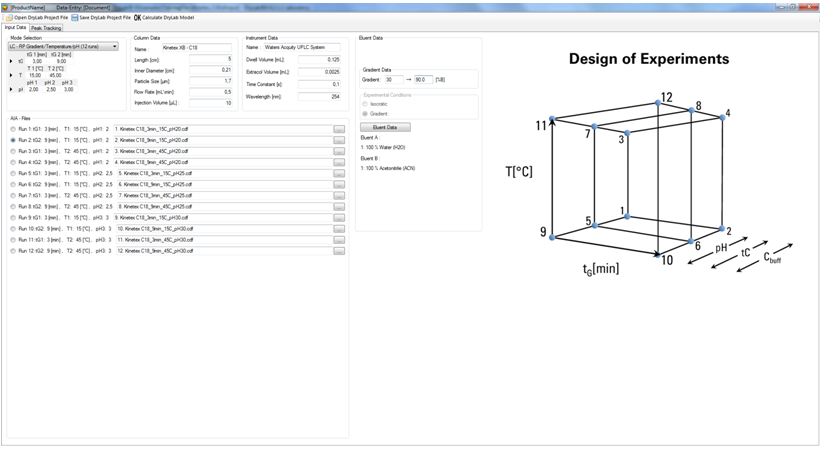
Using DryLab® to shape initial Design of Experiment (DoE)
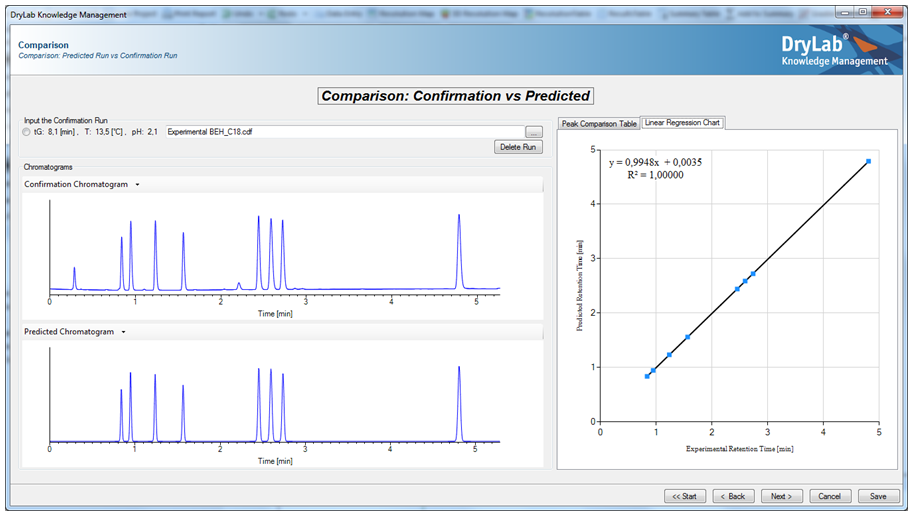
DryLab® Knowledge Management Module generates comprehensive cGMP method report
Related Articles










